Миеломенингоцеле поясничного отдела позвоночника

Менингомиелоцеле – это грыжа спинномозгового канала, при которой происходит выпячивание тканей и вещества спинного мозга через костный дефект позвоночного столба. Клиническая картина включает в себя наличие грыжеобразного выпячивания на спине ребенка в поясничной или крестцовой области. Сразу или с возрастом возникает нарушение иннервации нижерасположенных сегментов, вследствие чего развиваются тазовая дисфункция, парапарезы или параплегия. Диагностика основывается на наружном осмотре, подтверждении поражения ЦНС при помощи КТ и МРТ. Лечение хирургическое с последующей симптоматической терапией.
Общие сведения
Менингомиелоцеле или spina bifida cystica – это одна из форм спинального дизрафизма, которая проявляется выходом тканей спинного мозга за пределы позвоночного канала через костный дефект дужек. Впервые патология была описана в 1641 году Н. Тульпиусом. Распространенность составляет 5-20 на 10000 новорожденных. Является тяжелым заболеванием, которое вызывает серьезные неврологические нарушения и в некоторых случаях приводит к полному обездвиживанию больного. При менингомиелоцеле грыжевой мешок содержит оболочки спинного мозга, спинномозговую жидкость и корешки спинальных нервов. Чаще заболевание наблюдается при беременности матери в возрасте после 35 лет. В 6-8% случаев прослеживается наследственная склонность, что свидетельствует о генетическом характере патологии. При адекватном своевременном лечении прогноз относительно благоприятный.

Менингомиелоцеле
Причины менингомиелоцеле
Менингомиелоцеле развивается при наличии одного или нескольких факторов риска со стороны матери: прием фармакологических средств (оральные контрацептивы, препараты из групп салицилатов, вальпроатов, амфетаминов); употребление алкогольных напитков, табачных изделий, наркотиков, других тератогенных веществ; недостаточность микроэлементов и питательных веществ (в частности – Zn, Fe, фолиевая кислота) в рационе беременной; TORCH-инфекции (чаще всего – вирусы краснухи, гриппа и парагриппа). Также патология может иметь наследственный характер – установлен аутосомно-рецессивный механизм передачи заболевания, однако в 90-95% случаев семейный анамнез не отягощен.
Нервная трубка образуется из нервной пластинки на 19-20 день гестации. В нормальных условиях ее анатомическое закрытие происходит на 22-24 сутки, в результате чего остаются только верхнее и нижнее отверстие. Существует закономерность: чем позднее развивается менингомиелоцеле – тем ниже его локализация. Патогенез менингомиелоцеле досконально не изучен. На данный момент в педиатрии существуют 3 основных теории, объясняющих механизм развития данной патологии. Первая – это патология закрытия нервной трубки или теория Реклингхаузена. Она объясняет развитие спинального дизрафизма дефектом нейроэктодермы, возникающим в периоде раннего онтогенеза – на 25-30 день после оплодотворения.
Вторая – гидродинамическая или теория Моргани. Ее суть заключается в повышении давления внутри спинномозгового канала в I триместре беременности. Повышение давления провоцирует выпирание оболочек с последующим формированием костного дефекта и грыжи. Косвенным подтверждением данной теории является сопутствующая гидроцефалия в 90% случаев заболевания. Третья теория объясняет развитие менингомиелоцеле нарушением темпа роста тканей спинномозгового канала. При этом скорость дифференциации костных тканей позвоночного столба отстает или опережает развитие нервной трубки, из-за чего формируется дефект позвоночного столба.
Симптомы менингомиелоцеле
Клинические проявления менингомиелоцеле наблюдается уже с момента рождения ребенка. Основной признак – наличие характерного «мешка», который являет собой грыжеобразное выпячивание спинного мозга на спине ребенка. В некоторых случаях данное образование покрыто тонким шаром эпидермиса, но зачастую в дефекте позвоночного столба визуализируются непосредственно оболочки спинномозгового канала и нервные корешки. Наиболее характерная локализация – поясничный или крестцовый отдел. Примерно у половины новорожденных с изолированной формой изменения общего состояния изначально не возникает, однако с возрастом у всех детей появляются неврологические нарушения. Степень поражения напрямую зависит от уровня спинного мозга, на котором сформировалась грыжа.
При развитии менингомиелоцеле ниже 4 поперечного сегмента (L4) наблюдаются нарушения иннервации мочевого пузыря, реже – терминальных отделов кишечного тракта. Типичное проявление – недержание мочи. В более тяжелых случаях помимо тазовой дисфункции развиваются расстройства чувствительности и моторной функции нижних конечностей по типу парапареза. При локализации грыжевого выпячивания выше уровня 3 поперечного сегмента (L3) возникает полная параплегия, приводящая к тотальному обездвиживанию ног. Менингомиелоцеле зачастую сочетается с гидроцефалией и мальформацией Арнольда-Киари II типа с характерными для них клиническими проявлениями: увеличением размеров головы, рвотой, бессонницей, конвульсиями, атаксией, нарушением акта глотания, головными болями в области затылка, задержкой психофизического развития и т. д. Значительно реже в виде осложнения возникает эпилепсия.
Начиная с 12-ти месячного возраста, часто наблюдаются задержка в физическом развитии, набор излишней массы тела и деформации нижних конечностей. Ожирение в основном связано с малоподвижным или неподвижным образом жизни, а деформация – с отсутствием осевой нагрузки на ноги и отсутствием сопротивления для внутренних групп мышц стопы. На фоне дисфункции мочевого пузыря учащается пузырно-мочеточниковый рефлюкс, увеличивается вероятность развития инфекционных заболеваний мочевой системы, что может привести к почечной недостаточности.
Диагностика менингомиелоцеле
Диагностика менингомиелоцеле базируется на сборе анамнестических данных, проведении физикального обследования, использовании лабораторных и инструментальных методов исследования. При сборе анамнеза педиатр может установить факторы риска заболевания или возможную этиологию. Физикальное исследование заключается в непосредственном осмотре грыжевого выпячивания, определении уровня поражения и выявлении симптомов сопутствующих патологий. В общих лабораторных тестах (ОАК, ОАМ) при изолированной форме менингомиелоцеле отклонения от нормы не выявляются. При наличии в анамнезе данных, указывающих на возможную этиологию, могут использоваться специфические анализы – определение концентрации фолиевой кислоты, железа, цинка в плазме крови; ПЦР или ИФА на возбудителей TORCH-инфекций. Для уточнения уровня поражения спинного мозга и размера дефекта применяются методы нейровизуализации – компьютерная и магнито-резонансная томография. Также данные исследования позволяют выявить сопутствующие аномалии строения ЦНС – гидроцефалию, мальформацию Арнольда-Киари и другие. Дифференциальная диагностика менингомиелоцеле проводится с другой формой расщепления позвоночника – менингоцеле.
Лечение менингомиелоцеле
Основное лечение менингомиелоцеле осуществляется хирургическим путем. Суть – послойное закрытие дефекта и формирование спинномозгового столба. Ход операции: выделение и вскрытие грыжевого мешка, погружение тканей ЦНС в позвоночный канал, удаление дефекта и сшивание его остатков, коррекция дужек при помощи миофасциального лоскута. После операции, несмотря на восстановление нормальной структуры спинного мозга, избежать неврологических нарушений удается редко, т. к. ткани спинного мозга и корешков во время внутриутробного развития подвергаются необратимой дегенерации. При сопутствующей гидроцефалии также проводится ее нейрохирургическая коррекция.
Симптоматическая терапия подразумевает лечение развившихся осложнений. Назначают уросептики при частых инфекциях мочеполовой системы, оксибутинина гидрохлорид при дисфункции мочеиспускания и антихолинергические средства для стимуляции нейромедиаторной передачи. Важная роль отводится коррекции рациона ребенка, направленной на компенсацию дефицита микроэлементов. Применяют препараты цинка и железа, фолиевой кислоты, витамина С и В12. При частых запорах рекомендовано увеличить объем потребляемой жидкости.
Прогноз и профилактика менингомиелоцеле
Прогноз при менингомиелоцеле зависит от эффективности проводимого лечения. Как правило, при своевременной хирургической коррекции патологии, адекватной симптоматической терапии и рациональном питании исход достаточно благоприятный.
Специфической профилактики для данной патологии не существует. При отягощенном семейном анамнезе беременной проводится амниоцентез с целью антенатальной диагностики дефектов строения позвоночного столба и спинного мозга плода. Кроме того, используются общепринятые методы исследования в период беременности: УЗИ, определение концентрации альфа-фетопротеина в околоплодных водах. При выявлении TORCH-инфекций осуществляется их лечение и полноценное обследование матери и плода после проведенной терапии. Беременным рекомендуется увеличить количества метионина, витамина В12 и фолиевой кислоты в рационе, т. к. данные вещества снижают риск расщепления позвоночника ребенка.
Источник
Spina bifida — врожденный порок развития, связанный с нарушением формирования эмбриональной нервной трубки или смыкания ее краев и сопровождающийся серьезными неврологическими нарушениями, такими как паралич или нарушение функции органов малого таза.
По распространенности среди всех аномалий развития занимает второе место после врожденных пороков сердца, а самым частым вариантом дефекта нервной трубки является миеломенингоцеле. Новые методы лечения позволяют улучшить прогноз и качество жизни детей с данным пороком развития. Одним из таких методов является внутриутробное хирургическое закрытие дефекта.
Патогенез
В норме заращение нервной трубки происходит на 3–4 неделе эмбрионального развития. В формировании нервной трубки выделяют два этапа: первичная нейруляция и вторичная. Первичная нейруляция — процесс сращения краев нервной пластинки, вторичная нейруляция — формирование ее нижних отделов.
Рисунок 1 | Схема формирования нервной трубки
Воздействие тератогенных факторов, недостаток фолиевой кислоты или хромосомные аномалии могут нарушать процесс первичной и вторичной нейруляции, что приводит к образованию различных вариантов spina bifida: бессимптомной формы, связанной с недостаточным сближением позвонков (spina bifida occulta), менингоцеле и миеломенингоцеле (spina bifida cystica).
Рисунок 2 | Классификация дефектов нервной трубки
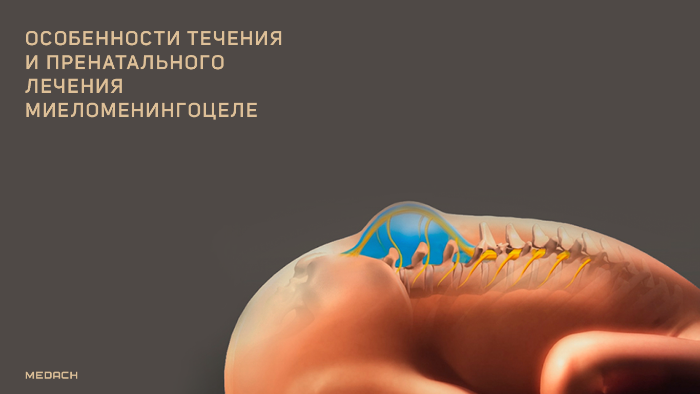
Миеломенингоцеле является наиболее тяжелой формой заболевания и сопровождается выходом в грыжевой мешок мозгового вещества и мозговых оболочек. Дефект может располагаться в любом месте вдоль оси позвоночника, но чаще всего находится в поясничном отделе. Миеломенингоцеле также связано с неправильным развитием краниального конца нервной трубки, что приводит к нескольким характерным аномалиям ЦНС.
Для мальформации Киари III типа характерна гипоплазия мозжечка и различная степень каудального смещения нижних отделов ствола головного мозга в шейный отдел позвоночного канала через большое затылочное отверстие. Эта деформация препятствует движению и абсорбции спинномозговой жидкости и вызывает гидроцефалию, которая встречается у более чем 90 % младенцев с миеломенингоцеле. Также у детей наблюдаются психоневрологические нарушения, паралич нижних конечностей, нейрогенная дисфункция мочевого пузыря и кишечника, сопровождающаяся недержанием.
Рисунок 3 | Новорожденный с миеломенингоцеле
На ранних сроках гестации при гистологическом исследовании тканей спинного мозга из области незавершенной нейруляции, изменения, как правило, не выявляются. Патологические изменения, такие как эрозии, кровоизлияния, воспаление или дистрофия возникают с увеличением срока гестации. Причиной может быть токсическое действие веществ, растворенных в амниотической жидкости, кроме того, содержимое грыжевого мешка может значительно повреждаться во время родов.
Таким образом, в патогенезе неврологических нарушений при миеломенингоцеле можно выделить два основных фактора: незавершенная нейруляция и повреждение тканей вследствие травм, воспаления и дегенеративных изменений (так называемый «двойной удар»). На основании этих данных можно сделать вывод, что заболевание прогрессирует во время беременности.
Преимуществом пренатального лечения является предупреждение такого прогрессирования патологии, а значит, уменьшение негативного влияния на ЦНС «двойного удара» и улучшение прогноза. Тщательное пренатальное обследование и раннее закрытие дефекта способствуют улучшению качества жизни детей и уменьшению частоты осложнений.
Сущность оперативного лечения
Операцию рекомендуется проводить между 19 и 25 неделями гестации. Беременная женщина, вынашивающая ребенка с диагнозом миеломенингоцеле, должна пройти ряд исследований для оценки возможности оперативного вмешательства:
- акушерское обследование;
- УЗИ плода с целью определения функции нижних конечностей (в т. ч. деформации стопы), а также уровня, на котором расположен дефект позвоночника;
- генетический скрининг;
- эхокардиография плода с целью выявления пороков сердца;
- МРТ с целью определения мальформации Киари II типа, грыжи заднего мозга и других аномалий развития головного мозга, в том числе гидроцефалии.
Критерии для проведения операции:
— возраст матери старше 18 лет;
— срок гестации между 19 и 25 неделями;
— нормальный кариотип плода;
— расположение грыжевого мешка в районе S1 и выше;
— подтвержденное с помощью УЗИ и МРТ выпячивание заднего мозга.
Противопоказания:
— многоплодная беременность;
— наличие других аномалий развития совместно со spina bifida;
— фетальный кифоз 30 градусов и более;
— предлежание плаценты;
— мягкая и/или короткая шейка матки (< 20 мм по результатам УЗИ);
— спонтанные ранние роды в анамнезе (первые роды в сроке < 37 недель);
— резус-конфликт;
— гестационный сахарный диабет;
— ожирение с ИМТ 35 и более;
— ВИЧ, гепатит В, гепатит С;
— аномалии развития матки;
— другие серьезные заболевания матери;
— психосоциальные ограничения;
— ограничение передвижения.
Соблюдение данных критериев позволяет снизить негативные последствия операции как для матери, так и для плода.
Операция проводится в два этапа:
- Нижняя поперечная лапаротомия, вертикальная гистеротомия с наложением скоб;
- Закрытие дефекта у плода.
Рисунок 4 | Методика проведения операции
Операция проводится под общим наркозом, обеспечивающим одновременное обезболивание матери и плода. Киста иссекается, дефект послойно ушивается и закрывается с помощью сшивания боковых кожных лоскутов, что формирует0 защиту развивающегося спинного мозга.
Рисунок 5 | Техника послойного ушивания дефекта
К послеоперационным осложнениям можно отнести хориоамнионит, сверхранние роды (до 28 недель), маловодие вследствие потери амниотической жидкости. Учитывая риск данных осложнений, требуется контроль состояния матери и плода.
Заключение
Принимая во внимание тот факт, что патологические изменения в спинном мозге у пациентов с миеломенингоцеле нарастают с увеличением срока гестации, пренатальная коррекция данного порока развития будет оптимальным вариантом, позволяющим достоверно снизить риск развития паралича, дисфункции мочевого пузыря и кишечника и других расстройств, следовательно, улучшить качество жизни пациентов.
Источники:
- Spina Bifida [Электронный ресурс] / Mark R Foster, MD, PhD, FACS et al. — Электрон. журн. — 2018. — Режим доступа: https://emedicine.medscape.com/article/311113-overview, свободный
- N Scott Adzick, Leslie N Sutton, Timothy M Crombleholme, Alan W Flake. Successful fetal surgery for spina bifida// The Lancet, Vol 352, 1998
- Andrew J. Copp, N. Scott Adzick, Lyn S. Chitty, Jack M. Fletcher, Grayson N. Holmbeck, Gary M. Shaw. Spina bifida // Nature Reviews Disease Primers, Vol.1, 2015
- N.S. Adzick. Fetal surgery for spina bifida: Past, present, future. // Seminars in Pediatric Surgery Vol.22, 2013
- А.С. Иова, Ю.А. Гармашов, Е.Ю. Крюков, Д.А. Иова. Возможности и перспективы пренатальной нейрохирургии (Нейрохирургии плода). // Вестник Северо-Западного государственного медицинского университета им. И.И. Мечникова. Том 7, №2, 2015
- Luc Joyeux,Enrico Danzer, Alan W Flake, Jan Deprest. Fetal surgery for spina bifda aperta //Child Fetal Neonatal Ed, 2018
Нашли опечатку? Выделите фрагмент и нажмите Ctrl+Enter.
Источник
Миеломенингоцеле. Причины и клиникаМиеломенингоцеле представляет собой наиболее тяжелую форму дизрафии с поражением позвоночника и возникает с частотой примерно 1:4000 родившихся живыми. Существуют надежные доказательства, что у матерей, получающих заместительную терапию фолиевой кислотой до зачатия, риск пороков развития нервной трубки снижается на 50 %. Заместительная терапия фолиевой кислотой эффективна в случае ее начала до зачатия и продолжения по крайней мере до 12-й недели беременности, т. е. до завершения нейруляции. Служба здравоохранения США рекомендует всем женщинам репродуктивного возраста, способным к деторождению, принимать фолиевую кислоту в дозе 0,4 мг/сут, а женщинам, у которых во время предшествующих беременностей зарегистрировано формирование дефектов нервной трубки плода, — по 4 мг ежедневно, начиная прием препарата за 1 мес. до начала планируемой беременности. Современная диета обеспечивает примерно 1/2 суточной потребности в фолиевой кислоте. С целью увеличения содержания фолиевой кислоты в пищевом рационе в 1998 г. в США и Канаде выпускались пищевые продукты, обогащенные фолиевой кислотой: мука, макаронные изделия, рис и кукурузная мука содержали 0,15 мг фолиевой кислоты на 100 г продукта. Но, к сожалению, доза фолиевой кислоты, получаемой с пищей, не достигает минимальной дозы, позволяющей предотвратить пороки развития нервной трубки. Поэтому необходимо создание информативных обучающих программ для женщин, планирующих беременность. Некоторые лекарственные препараты, включая антагонисты фолиевой кислоты, такие как триметоприм и антиконвульсанты (фенитоин, карбамазепин, фенобарбитал и примидон), повышают риск развития миеломенингоцеле. При приеме антиконвульсантов — препаратов вальпроевой кислоты во время беременности риск пороков развития нервной трубки достигает примерно 1-2 %. Некоторые врачи рекомендуют всем женщинам репродуктивного возраста, страдающим эпилепсией и принимающим антиэпилептические препараты, получать заместительную терапию фолиевой кислотой. Миеломенингоцеле заболевание вызывает дисфункцию не только периферической и центральной нервной системы, но и многих других органов и структур организма, включая костную систему, кожу, мочеполовую систему. Миеломенингоцеле (спинномозговая грыжа) может локализоваться в любом участке вдоль нервной трубки (осевой части ЦНС), однако наиболее часто (75 % случаев) локализуется в пояснично-крестцовом отделе. Распространенность и степень тяжести неврологического дефицита зависят от локализации миеломенингоцеле.
Поражение в нижней части крестцовой области приводит к нарушению функции тазовых органов (недержание мочи и кала) в сочетании с анестезией в области промежности, но без изменения двигательной функции. Миеломенингоцеле, локализующееся у новорожденных в средней части крестцовой области, обычно имеет мешковидную форму, кистообразную структуру и покрыто тонким слоем частично эпителизированной ткани. Под оболочкой видны рудименты нервной ткани, возможен разрыв оболочки с истечением ликвора. При неврологическом осмотре выявляются вялый паралич нижних конечностей, отсутствие глубоких сухожильных рефлексов, реакции на прикосновение и болевой чувствительности; с высокой частотой встречаются постуральные аномалии нижних конечностей, включая косолапость и подвывих бедер. Возможно постоянное выделение мочи по каплям и расслабление анального сфинктера. Таким образом, при локализации миеломенингоцеле в среднем поясничном отделе отмечаются признаки поражения периферических мотонейронов вследствие поражения конуса спинного мозга. При распространении миеломенингоцеле на грудной отдел спинного мозга, как правило, выявляются прогрессирующие неврологические нарушения. Тем не менее у пациентов с миеломенингоцеле в верхней грудной или шейной области обычно имеется минимальный неврологический дефицит и в большинстве случаев гидроцефалия не развивается. Гидроцефалия в сочетании с аномалией Киари типа II развивается по крайней мере в 80 % случаев у пациентов с миеломенингоцеле. Как правило, чем ниже локализовано миеломенингоцеле (например, в крестцовом отделе), тем меньше риск гидроцефалии. Увеличение желудочков может быть медленно прогрессирующим и не проявляться клинически или развивается быстро, приводя к внутричерепной гипертензии (выбухание переднего родничка, дилатация черепных вен, симптом «заходящего солнца», раздражительность и рвота в сочетании с увеличением окружности головы). Нередко у младенцев с гидроцефалией и аномалией Киари типа II выявляются симптомы дисфункции ромбовидного мозга, включая нарушение глотания, поперхивание, стридор, апноэ, паралич голосовых связок, слюнотечение, а также спастичность в верхних конечностях. В отсутствие терапии возможен летальный исход. Такие кризисы Киари обусловлены вклинением продолговатого мозга и миндалины мозжечка в большое затылочное отверстие. — Также рекомендуем «Лечение миеломенингоцеле. Прогноз» Оглавление темы «Неврологические нарушения у детей»:
|
Источник